
Improving the last prediction
In my first post I showed how to build a simple model, using only genetic information, that predicts height with 53% accuracy in an independent sample. In this post I’m going to improve that model, which will ultimately result in a model with 64% accuracy in an independent sample – an 11% improvement! It’s also slightly simpler – only 4 variables in the regression, instead of 5.
The bulk of the improvement comes from something fairly straightforward – data cleaning. Using just PCA and k-means clustering I identify sources of population stratification and genotyping chip effects. Removing these people from the model (N=164) improves the accuracy by a whopping 9%. As a fun aside, I also show that the model is particularly inaccurate in participants who are descended from ancestral populations other than the one used for the GWAS (in this case non-Europeans), and that the error is right-skewed. This is, of course, why cross-ancestry genetic prediction is so hard to do.
In the last step, I use a fun new method – MTAG – to run a meta-analysis of two GWAS for height. The new genetic risk score improves on my original one by an additional 2% (about the amount one would expect).
Finally, I have to give a shout-out to the folks over at crowdAI for taking the time to assemble this great data set, and also for being kind enough to give me additional information (like the participant’s OpenSNP ids, and the heights for the test data-set) even though they haven’t made it publicly available yet (I assume it will be posted soon enough). And of course, an enormous amount of thanks goes to the folks at openSNP, who were willing to put their genome into the public domain.
Ancestry is the real kicker
Population stratification is real, and it will interfere with any genetic analysis that doesn’t try to account for it. It manifests as, among other things, differences in allele frequency. Individual genetic mutations have very small effects, so most of the time, if a mutation is more frequent in one population than another, it doesn’t actually mean anything (e.g. it doesn’t mean that either population necessarily has greater genetic risk for an associated disorder). But, if you’re doing an analysis with a lot of variables, and many of them have different distributions across large sub-groups of your sample, then your results are definitely going to be hurt (e.g. increased Type I and Type II errors). Probably what you would want to do is repeat the analysis in each individual group, or if they’re too small, remove the small ones. Here I’m going to remove people from the analysis, as the vast majority of the sample is of European ancestry.
Ancestry is going to affect the analysis in two ways. First, it’s a multi-ancestry data set, so ancestry will increase the error of the model-fit, simply because there will be a subset of the participants who are different from the majority (which are of European ancestry). Second, the model uses a genetic risk score that is derived from an independent study of Height that was conducted in participants of European ancestry. As a result, analyses in non-European ancestry groups will have increased error, and will be biased by differences in allele frequency.
Thanks to the information from the crowdAI folks, I was able to look at the other data, besides height, that is available for the OpenSNP participants. A subset of 250 of them responded to at least one question about their ethnicity or ancestry. Of these, 228, or 91%, responded that they were of European ancestry. I’m going to use this information as a sanity-check, to confirm my intuition about which participants are of European-ancestry.
Finding and removing population stratification
Some basic steps for dealing with population stratification. If you’re doing this in a real analysis that you care about, I would strongly suggest using something more rigorous, such as EIGENSOFT.
- PCA of the genetic data
- Scree plot identifies 3 PCs before the elbow
- K-mean clustering scree plot identifies 4 clusters before the elbow

So this looks like a fairly standard plot of PCs of genetic data. Since the sample is mostly of European ancestry, we expect the clusters closest to [0,0,0] to be those participants. The shape of clusters 2 & 3 (the two smaller ones) is also consistent with what I’ve seen African and Asian ancestry clusters look like in other data sets. Finally, the self-report data confirms that the participants who say they’re of European ancestry are all in clusters 1 & 4, with only 1 exception. So we remove those two clusters.
But is this a good idea? I’m basically throwing away data! So, I ran the same model as in my original solution (Height ~ Sex + PC1 + PC2 + PC3 + PRS-Height) [PRS-Height = Polygenic Risk Score, or Genetic Risk Score, for height], but with leave-one-out cross validation. Here’s a plot comparing the distribution of the error (i.e. the difference between predicted and true height) between the people who I’m going to remove, and those who I’ll keep.
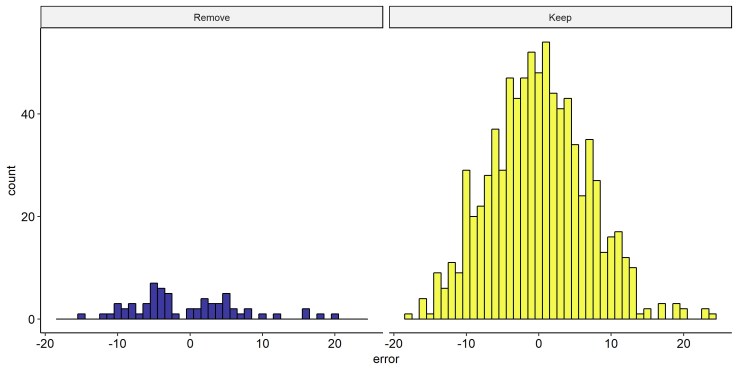
We see that the distributions certainly look somewhat different, though the ‘Keep’ group is so much larger, it’s hard to tell. Comparing the statistics of the distributions, the ‘Remove’ group has a slightly larger skew (0.66 vs 0.2) and standard-deviation (7.4 vs 6.7), but on the face of it, it isn’t clear that it’s so different. So I want to test whether the ‘Remove’ group really is different. To do this I drew random samples of N=64 (the size of the ‘Remove’ group) from the ‘Keep’ group, with replacement. Doing this 2 million times (overkill), I compare how likely it is for a sample of N=64 drawn from ‘Keep’ distribution to have the Skew and SD of the ‘Remove’ group.
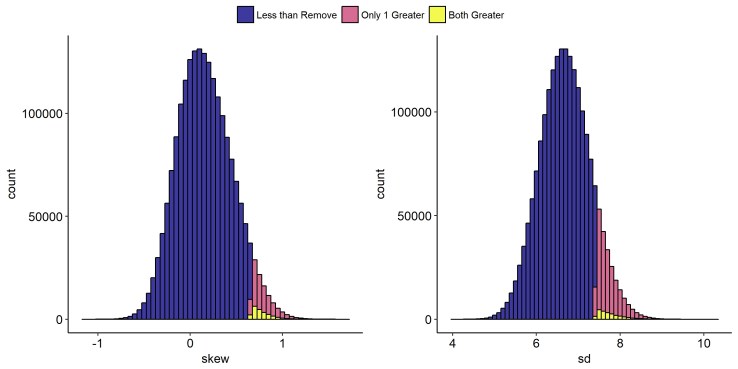
From this we see that it’s fairly unlikely that the ‘Remove’ group error is drawn from the same distribution as the ‘Keep’ group error. While either the Skew or the SD of the ‘Remove’ group is reasonable (the red bands in the histogram), only in 1.12% of samples does a random draw from the ‘Keep’ group have both a higher SD and Skew than the ‘Remove’ group (i.e. the yellow bands). So it’s clear that the ‘Remove’ group has higher error, and that the error is right-skewed. For more information, see this paper, which covers this sort of bias is much greater detail.
Finding and removing batch & chip effects
Batch effects are a problem. Essentially, variability between samples, either in terms of the technology used (which chip), or the calling algorithm, or the criteria used to make calls, will increase errors in any analysis that uses the data. We can use the same steps as above to see if there’s evidence of batch-effects (NB there are lots of very sophisticated ways to do this, I’m just showing a basic approach).
- PCA of the genetic data
- Scree plot identifies 3 PCs before the elbow (again)
- K-mean clustering scree plot identifies 4 clusters before the elbow (again)

Are these just different sub-populations? Northern/Southern or something? We can check which company genotyped each person, and see if there are differences (obviously not perfect, since most companies have transitioned through multiple chips over the years). Looking at this, it’s clear that there is a chip effect, as cluster 3 consists exclusively of the majority of the people genotyped by Family Tree DNA (70 people in that cluster). It it’s that easy to tell which chip someone was genotyped on, then it almost certainly will affect the analysis. So I’ll remove that cluster, and with those people removed, only 25 people not genotyped by 23andME (the most popular company, apparently) remain. For safety’s sake, I’m removing all of them.
Let’s do an analysis!
- PCA of the genetic data
- Scree plot identifies 2 PCs before the elbow
- Repeat the same analysis as before
Now there are N=623 in the training data, and N=105 in the test data. There are 4 variables in the linear model:
- Chromosomal sex (still explaining the VAST majority of variance)
- Principal components of ancestry (x2)
- Polygenic Risk Score for ancestry
In the training data, the model explains a full 60% of the variance. I then used the model to predict height in the test data, with 62% accuracy. Like I said, that’s a 9% improvement over my last model. It’s nuts, but population stratification & chip effects really do have a big effect!
Improve the model even more
But can we do better? Recently, a super-cool approach, called ‘Multi-Trait Analysis of GWAS, or MTAG was published. I decided to have some fun and improve my model some more using this method. Using MTAG, I meta-analyzed the results from the GWAS that went into my initial risk score, with GWAS results for Height from the UK Biobank Study, which have been compiled and released by Ben Neale’s group (N=336,000). The result was GWAS summary statistics from the equivalent of a sample of N=459,000. I reran my model with a polygenic risk score derived from this improved GWAS, which resulted in test-set accuracy of 64%, or a 2% improvement. This is right on par with what one would expect, based on the prior work using this approach and related techniques. In this final model, data gleaned from genetic variation improves the prediction of height by 21% over just chromosomal sex. This is about the amount we would expect.
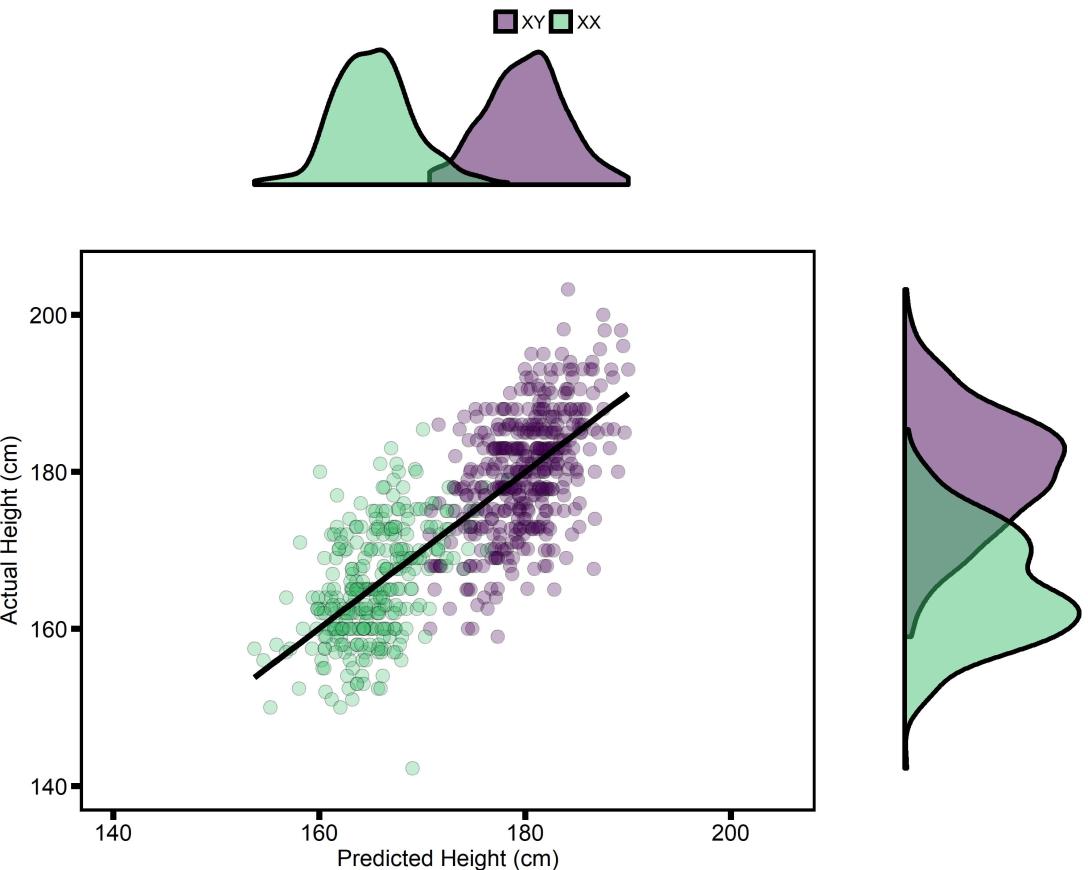
Finally, to plot the results of the final model, I used leave-one-out cross validation to compute height-predictions for all 728 of the participants. I plot them below, grouped by chromosomal sex (which I would note is still far and above the strongest variable in the model).

The points are colored by chromosomal sex. The density plots make it clear that, while a majority of the variance explained is coming from chromosomal sex, the genetic risk score adds a reasonable amount of information on top. Additionally, while this is quite good performance, there is still a ton of error! So while genetics proved useful information, especially in a research context, I wouldn’t recommend going and taking this sort of model too seriously yet.

[…] If you’re reading this, you may also be interested in the follow-up post, where I further improve this model by an additional […]
LikeLike